Navigation
Navigation
Sie sind hier:
Startseite
![]() Projekte
Projekte
![]() Blue Genes
Blue Genes
Seiteninhalt
![]()
„Der Name ,Blue Genes‘ macht deutlich, was im Experiment passiert: Bakterien färben sich durch die Übertragung eines Genfragmentes blau. Schüler und Lehrer werden durch dieses Experiment mit den grundlegenden Techniken der Molekularbiologie vertraut und arbeiten wie die Wissenschaftler im Labor. Der Genbaukasten ,Blue Genes‘ ist Wissenschaft zum Anfassen.“ (Roche/Blue Genes)
Tag (21.02.06)
Schneiden des Plasmids pUCD-lacZ mit dem Restriktionsenzym Ban II
Zunächst schneiden wir ein Plasmid (eine ringförmige DNA) mit einem Restriktionsenzym, welches an uns bekannten Stellen schneidet.
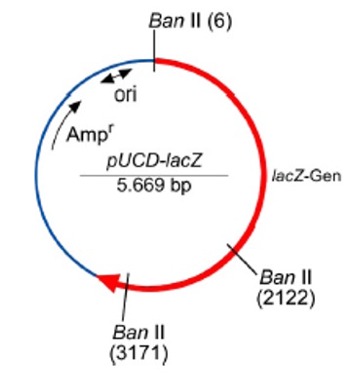
Abb: Schematische Darstellung des Plasmids pUCD-LacZ (Quelle: Roche/Blue Genes)
Vorbereitung des Agarose-Gels
Für die Elektrophorese benötigen wir einen Elektrophoresepuffer, damit eine elektrische Spannung gewährleistet wird. Außerdem braucht man ein Agarose-Gel, in welches die Proben pipettiert werden.
Das Agarose-Gel wird in eine Elektrophoresekammer gegossen und es werden Kämme für die Entstehung der Slots eingesetzt.
Vorbereitung der Proben für die Elektrophorese
Für die Elektrophoresevorbereitung benötigen wir drei Proben: den DNA-Marker, die Kontrolle und den Restriktionsansatz vom Anfang. Der DNA-Marker enthält bestimmte DNA-Fragmente, deren Basenpaarlängen uns bekannt sind. Die Kontrolle enthält das ungeschnittene Plasmid pUCD-lacZ. Der Restriktionsansatz enthält das voraussichtlich geschnittene Plasmid.
Alle drei Proben enthalten außerdem einen Auftragspuffer. Dieser hat an sich eine hohe spezifische Dichte. Somit können die drei Proben im Agarose-Gel sinken und letztendlich nicht heraus diffundieren. Da der Austragspuffer noch Farbe enthält, kann man die Wanderung der DNA während der Elektrophorese beobachten.
Tag (23.02.06)
Elektrophorese

In das Agarose-Gel in der Elektrophoresekammer werden die einzelnen Proben pipettiert und, nachdem die Kammer mit dem Puffer übergossen wurde, eine Spannung angelegt. Das Agarose-Gel wirkt wie ein Sieb, indem kleine DNA-Fragmente wesentlich schneller und weiter im Gel laufen als große. Durch Vergleichen mit den anderen Proben kann man die einzelnen Längen der DNA-Fragmente bestimmen.
Färben der DNA im Agarose-Gel

Um die Banden der DNA-Fragmente sichtbar zu machen, benötigen wir eine Farblösung aus AzurB-Chlorid. Um das Agarose-Gel wieder zu entfärben, spült man das Gel mit destilliertem Wasser. Nach ungefähr 10 Minuten sollte man die Banden im Agarose-Gel erkennen können.
Im Allgemeinen ist ein insgesamt erfolgreiches Verlaufen der jeweiligen Spuren zu erkennen und der Verlauf der Banden in Spur 3 lässt auf ein gelungenes Schneiden der DNA-Fragmente schließen.
In Spur 1 ist der DNA-Marker zu sehen, in Spur 2 das ungeschnittene Plasmid, welches in den zwei Konformationen supercoiled und zirkulär vorliegen kann, in Spur 3 erkennt man drei Banden, die auf ein erfolgreiches Schneiden des Plasmids pUCDlacZ schließen lassen.
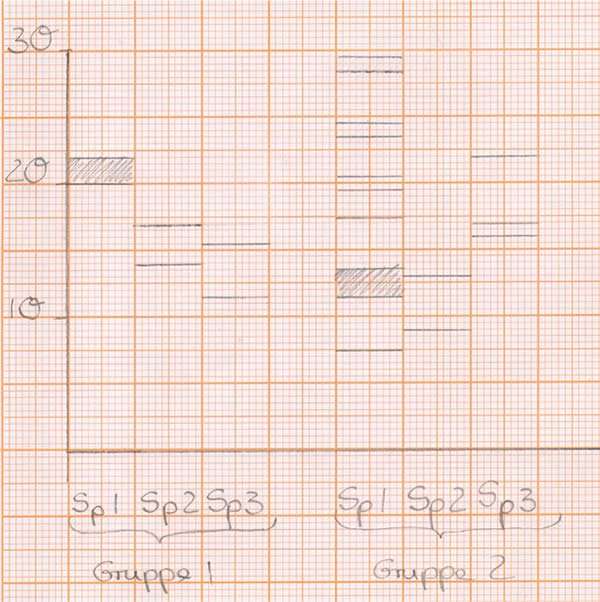
Abb: Gel 1

Abb: Gel 2
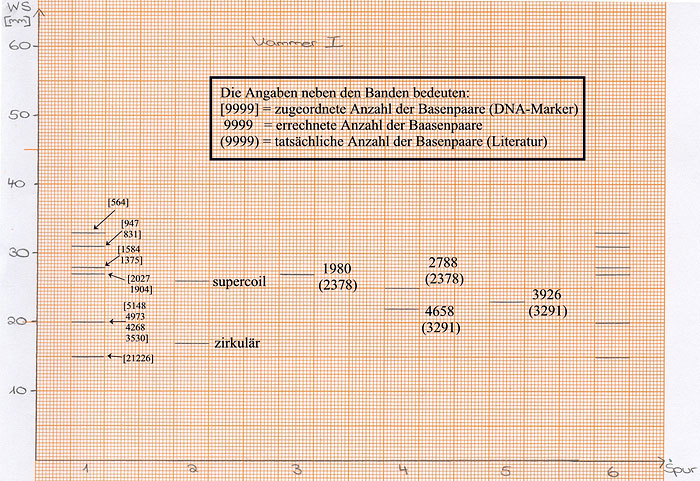
Um die Größe der geschnittenen Fragmente abschätzen zu können, ist es notwenig eine Eichkurve zu erstellen.
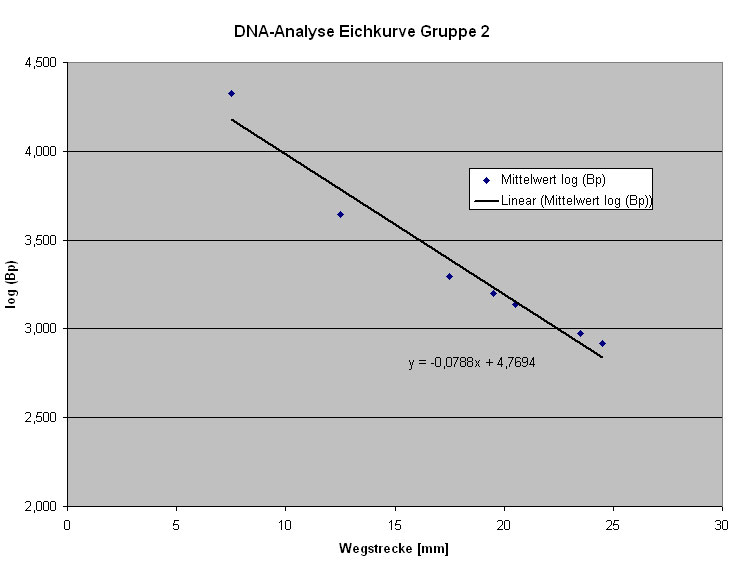
Abb: Eichkurve
Die Eichkurve ergibt sich aus der Auswertung der Banden des DNA-Markers, in dem man die (z. T. gemittelt-) logarithmisierte Anzahl der Basenpaare gegen die Wegstrecke der Banden aufträgt.
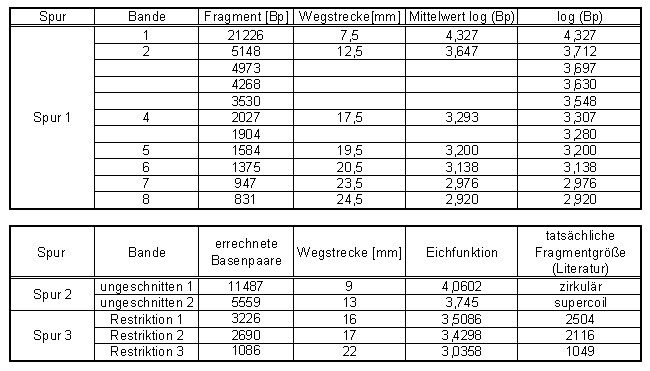
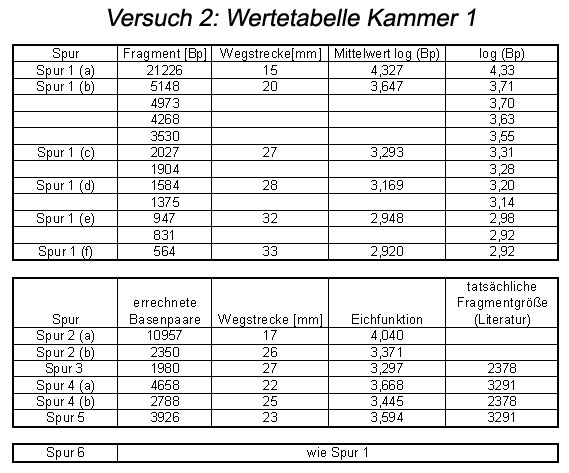
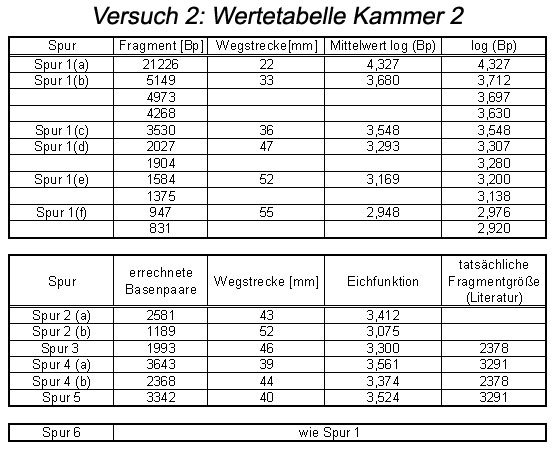
Abb: Wertetabelle
Aus der ermittelten Eichfunktion kann man dann die Anzahl der Basenpaare des eigenen Restriktionsansatzes ermitteln. Zur Versuchsauswertung eignet sich am besten das Ergebnis der Gruppe 2, an deren Gel sich die Banden des DNA-Markers am besten erkennen ließen.

Abb: Versuchsauswertung
Die Werte stimmen nicht genau mit denen der Literaturwerte überein, da ein Ablesefehler von 0,5mm bereits einen Unterschied von etwa 500 Basenpaaren ausmacht. Um abschließend eine genaue Anzahl zu ermitteln, wäre eine Sequenzierung notwendig.
Mögliche Gründe für die unterschiedlichen Ergebnisse:
(Junker, Kramer, Saul)
Im ersten Schritt pipettiert Gruppe 1 das pUCD-Plasmid in ein Eppendorfgefäß und gibt dann die Restriktionsenzyme Bam H I und Hin D III hinzu. Gruppe 2 setzt gleichzeitig einen Kontrollversuch mit der Plasmid pUCD-LacZ-DNA an, mit dem das korrekte Schneiden und Einsetzen des Plasmids LacZ überprüft werden soll.
Die Restriktionsansätze werden nun durch ein 37°C warmes Wasserbad aktiviert und die Reaktion anschließend mit einem 65°C heißem Wasserbad beendet. Danach werden die Produkte bis zur weiteren Verwendung auf Eis gelagert.
Nun wird die Elektrophorese vorbereitet. Zusätzlich zu den Restriktionsansätzen werden Kontrollen laufen gelassen: Bei jeder Gruppe zur Skalierung ein DNA-Marker und bei Gruppe 1 zusätzlich ungeschnittene pUCD-DNA und bei Gruppe 2 das lacZ-Genfragment.
Gleichzeitig bereitet eine Gruppe schon die Agarplatten als spätere Nährböden für die Bakterien vor.
Nach der Kontrolle durch die Elektrophorese wird die Ligation d. h. das Einsetzen von dem lacZ-Genfragment, durch hinzufügen desselben zu dem geschnittenen pUCD-Plasmid, durchgeführt.
Nun werden die kompetenten Bakterien (E.coli K12 (JM 109)) langsam aufgetaut und erst der Ligationsansatz und dann die Nährlösung LB Medium hinzu gegeben.

Anschließend wird diese Lösung auf dem Agarboden unter sterilen Bedingungen verteilt und dann über Nacht bei 37°C in den Brutschrank gestellt. So werden die Bakterien mit dem Plasmid , also mit einer Ampicillin-Resistenz, selektiert und die Bakterien mit dem LacZ-Gen durch den Farbstoff X-Gal sichtbar gemacht. Es werden zudem vier verschiedene Kontrollansätze kultiviert. Diese enthalten:
(Fröhling, Knafla)
Zur Versuchsauswertung von Versuch 2 der Elektrophorese haben wir folgende Materialien miteinbezogen:
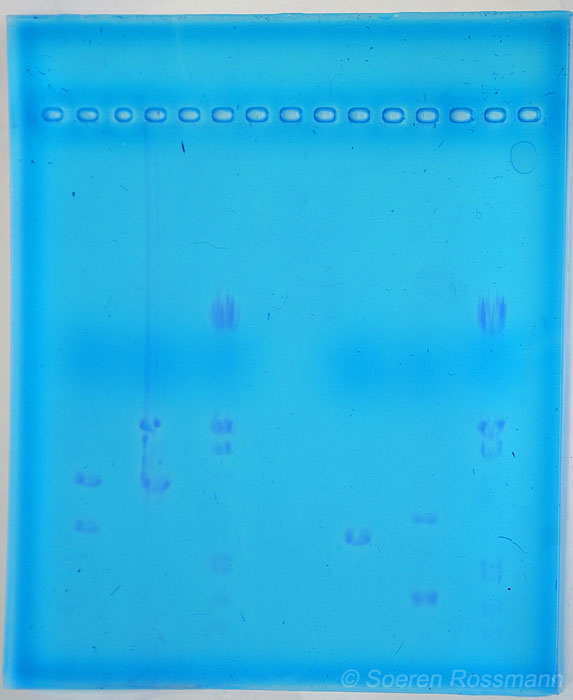

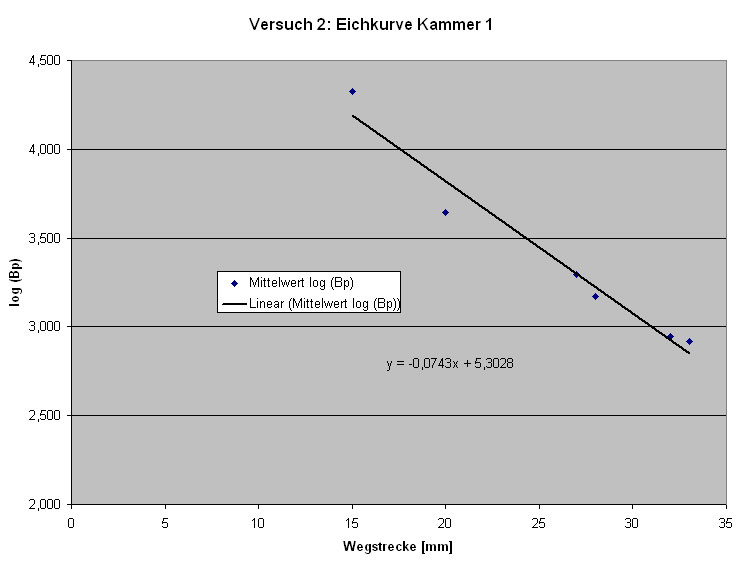
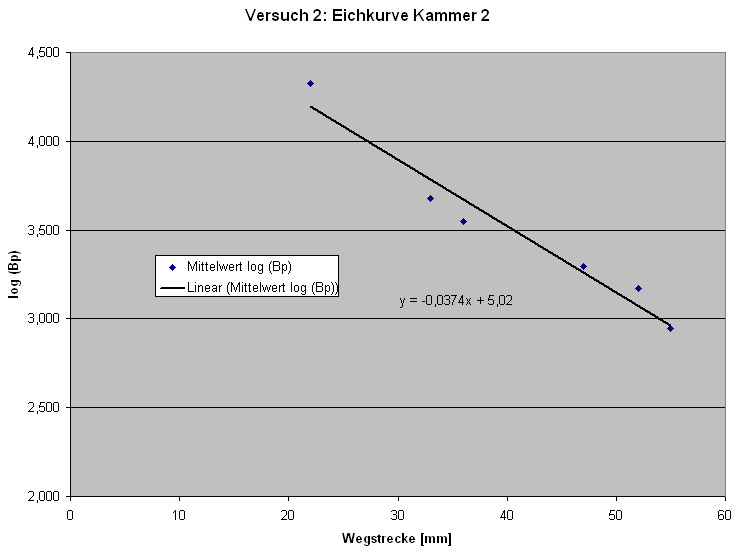
Für uns waren bei diesem Versuch mit der Elektrophorese die Spuren 2 und 3 von besonderer Bedeutung. Da in Spur 2 das ungeschnittene Plasmid aufgetragen wurde, welches in 2 Banden, zirkulär und supercoiled (siehe Spur 2) zu sehen ist und in Spur 3 unser Restriktionsansatz. Dieser wurde von den Enzymen Bam HI und Hin d III geschnitten, so dass man durch den Vergleich der beiden Spuren überprüfen kann, ob der Restriktionsansatz (Spur 3) geschnitten hat oder nicht.
Beide Kammern lieferten uns gute Ergebnisse. Wir haben die Auswertung exemplarisch an Kammer 1 durchgeführt.
Auf Spur 1 ist der DNA-Marker aufgetragen. Man erkennt hier sechs Banden, die verschiedene Fragmentgrößen darstellen. Der DNA-Marker dient zur Kontrolle der selbst ermittelten Werte auf den anderen Spuren. Die verschiedenen Längen der Banden kann man in der Grafik erkennen.
Auf Spur 2 wurde ein ungeschnittenes Plasmid aufgetragen. Das erkennt man daran, dass zwei Banden zu sehen sind. Eine Bande stellt die zirkuläre Form des Plasmids dar, welche größer ist und damit langsamer „läuft". Die andere Form ist „supercoiled“, sie ist kleiner und läuft daher schneller.
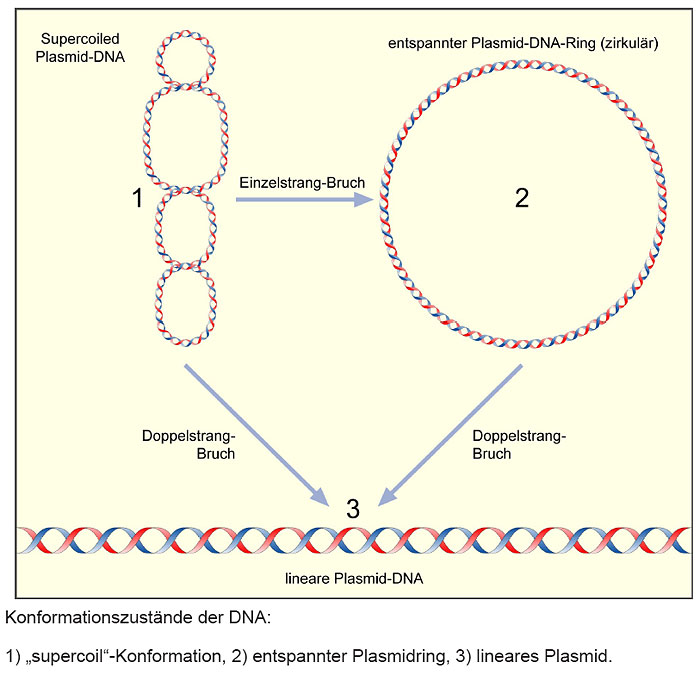
Abb: Konfirmationszustände der DNA (Quelle: Roche/Blue Genes)
In der Spur 3 ist der Restriktionsansatz, der mit den Enzymen Bam H I und Hin d III geschnitten wurde, erkennbar. Es liegt nur eine Bande vor, dies bedeutet, dass zweimal geschnitten wurde. Das kleinere Fragment beträgt nur 30 Basenpaare und ist daher nicht als Bande erkennbar.
Abb: Spur 3 (Quelle: Roche/Blue Genes)
In der Spur 4 ist ein anderer Restriktionsansatz mit pUCD-lacZ, der mit den Enzymen Bam H I und Hin d III geschnitten wurde, zu sehen. Daher sind hier 2 Banden erkennbar, eine für das geschnittene Plasmid wie in Spur 3 und eine für das lac-Z-Gen.
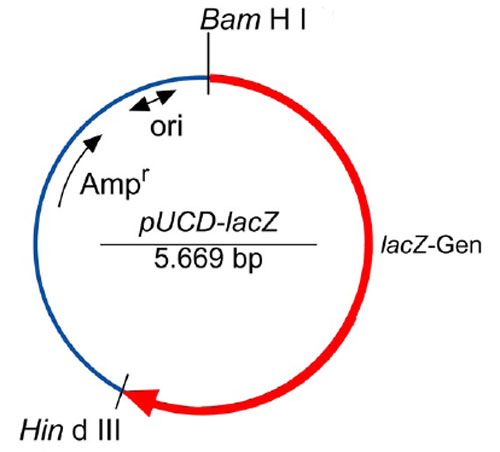
Abb: Spur 4 (Quelle: Roche/Blue Genes)
Auf der Spur 5 ist das lacZ-Gen aufgetragen, es ist nur eine Bande sichtbar. Diese lässt sich dadurch erklären, dass kein vollständiges Plasmid vorliegt, sondern nur das lacZ-Gen. Diese Spur dient zur Kontrolle der Spur 4, um das herausgeschnittene lacZ-Gen dort nachweisen zu können.
In der Spur 6 ist dann wieder, wie in Spur 1, der DNA-Marker zusehen. Auch hier zeigen sich sechs Banden mit verschiedenen Fragmentgrößen, die wiederum zur Kontrolle dienen.
Bei der Auswertung dieses Versuches kommt man durch den Vergleich der Spuren 2 und 3 zu dem Ergebnis, dass unser Restriktionsansatz (Spur3) geschnitten hat, da im Vergleich zu Spur 2 nicht mehr 2 Banden (zirkulär und supercoiled), sondern nur noch eine Bande erkennbar ist.
Im Folgenden werden wir die Ergebnisse unseres Versuchs betrachten. Die Analyse erfolgt anhand der Spuren die sich in unseren zwei Kammern ergaben:
Abb: Ergebnisse aus Gel 1
Abb: Ergebnisse aus Gel 2
Im Vergleich zu Kammer 1 kann man verschiedene Differenzen zu Kammer 2 feststellen. Es wurden jedoch in allen Spuren die gleichen Ansätze aufgetragen.
In Kammer 2 sind alle Fragmente deutlich weiter gelaufen als die Fragmente in Kammer 1. Das liegt wahrscheinlich daran, dass die beiden Gele eine unterschiedliche Konzentration aufweisen und so die unterschiedlichen Ergebnisse entstehen.
Unwahrscheinlicher ist, dass das Gerät der Kammer 2 die Stromübertragung nicht gleich gut gewährleistet oder durch einen Fehler in der Verkabelung die Stromfreisetzung vermindert wurde. Wahrscheinlicher ist hier jedoch, dass die Gele nicht identisch sind, dies kann schon durch kleine Abweichung in der Herstellung passieren.
Das Fragment in Spur 3 müsste identisch sein, zu dem Fragment in Spur 4 welches weiter gelaufen ist. Die geringe Abweichung die auf den Grafiken zu erkennen ist, wird durch geringe Ablesefehler entstanden sein oder dadurch, dass das Gel nicht an allen Stellen homogen ist.
Bei der Spur 2 in beiden Kammern kann man erkennen, dass diese unterschiedlich im Verhältnis zu dem zugehörigen DNA-Marker der jeweiligen Kammer steht. In Spur 2 wurde das ungeschnittene Plasmid aufgetragen und die beiden zu sehenden Banden werden durch die zwei verschiedenen Plasmidformen „zirkulär" und „supercoiled" bestimmt. Doch auch diese Formen können sich untereinander unterscheiden. Das heißt, dass zwei zirkuläre Formen unterschiedlich weit laufen, da sie nicht vollständig identisch sind. Genauso verhält es sich bei der "supercoiled" Form. So kann man die Abweichungen in den Verhältnissen der Kammern erklären.
Die Bande der Spur 3 müsste auf der Höhe des weiter gelaufenen Fragments in Spur 2 sein. Auch hier werden Ablesefehler und hier entstandene Ungenauigkeiten die Ursache der Abweichungen sein. So liegt in Kammer II die Bande der Spur 3 etwas zu niedrig und in Kammer I liegt die Bande der Spur 3 minimal unter dem vorgesehenen Wert.
(Malinowski, Sparding)
Erwartung: wenige blaue und weiße Bakterienkolonien
Ergebnis:
Deutung:
1. Gruppe: Transformation oder Ligation misslungen (siehe hierzu auch die folgenden Deutung).

Quelle: Roche/Blue Genes
Erwartung: viele blaue und weiße Bakterienkolonien
Ergebnis:
Deutung:
1. Gruppe: Transformation muss doch stattgefunden haben, da 2 Bakterienkolonien zu finden sind. Der Fehler liegt möglicherweise an einer falschen Verdünnung.

Quelle: Roche/Blue Genes
Erwartung: Koloniebildung von weißen Bakterien
Ergebnis: weißer Bakterienrasen; keine blauen Bakterienkolonien

Quelle: Roche/Blue Genes
Erwartung: keine Koloniebildung
Ergebnis: keine Koloniebildung

Quelle: Roche/Blue Genes
Erwartung: Koloniebildung von weißen Bakterien
Ergebnis: weißer Bakterienrasen; keine blauen Bakterienkolonien

Quelle: Roche/Blue Genes
Erwartung: Koloniebildung blauer Bakterien
Ergebnis: keine weißen Bakterienkolonien; 50 blaue Bakterienkolonien

Quelle: Roche/Blue Genes
(Gräper, Kröger)
Für unseren Blue-Genes-Versuch ist die Benutzung von Bakterien sinnvoll, da die Bakterien Fremd – DNA schnell aufnehmen können. Außerdem haben Bakterien die Eigenschaft, sich exponentiell zu vermehren.
Den Bakterien (E.-Coli) haben wir ein Plasmid, das aus einer ringförmigen DNA besteht. Das Plasmid kann zusätzliche Gene aufnehmen. Das Plasmid wird mit Hilfe von Restriktionsenzymen aufgeschnitten und mit Hilfe einer Ligase wird die Fremd-DNA in das Plasmid eingebunden. Das entstandene Plasmid wird in die Bakterienzellen transformiert. Damit die Transformation überhaupt möglich ist, muss die Zellwand der Bakterien durch Standartmethoden durchlässig gemacht werden. So erhält man so genannte kompetente Bakterien. Allerdings wird das Plasmid nur mit einer Wahrscheinlichkeit von 1:10.000 in die Zelle aufgenommen.
Unter optimalen Bedingungen können sich die Bakterien alle 20 Minuten durch Zellteilung verdoppeln. Dadurch kommt es zur exponentiellen Vermehrung, vorausgesetzt, alle Bakterien überleben. Bei unserem Versuch haben wir bereits im Labor speziell vorbereitete kompetente Bakterien benutzt. Ein optimales Wachstum haben wir durch das Nährmedium Agar und einer gleich bleibenden Temperatur von 37 °C im Brutschrank bei 24 Stunden erreicht.
Bei unserem speziellen Versuch kam uns zu Gute, dass der Transformationsansatz stark verdünnt vorlag. Somit bildeten sich nur wenige Kolonien, welche jeweils auf ein Bakterium zurückzuführen sind.
(Hildebrandt, Malec, Smolka)
Überall in der Luft befinden sich Bakterien und Mikroorganismen, die sich auf den Arbeitsgeräten absetzen können. Um dies zu vermeiden, müssen gewisse Vorschriften eingehalten werden. Dies soll dazu führen, dass wir möglichst genaue Ergebnisse erhalten und diese nicht durch Bakterien verfälscht werden.
Zum einen sollten Fenster und Türen geschlossen bleiben, damit starker Durchzug verhindert wird. Außerdem müssen die Arbeitsflächen sauber gehalten werden, nur die Arbeitsgeräte, die wirklich benutzt werden dürfen sich darauf befinden und die Geräte müssen bis zur Verwendung steril aufbewahrt werden. Besonders beachtet werden sollten:
Grundsätzlich sollten, besonders beim Pipettieren, Handschuhe getragen werden, um so vor zusätzlichen Verunreinigungen durch Bakterien zu schützen.
(Gräper, Kröger)

Abb: Eppendorfset (Quelle: IFM-GEOMAR
http://www.ifm.uni-kiel.de/OzeanOnline/
wiss_arbeit/rna-dna/rna.htm)
(Niemeyer, Mensah, Roßbach)












Auf einer weiteren Webseite gibt es noch mehr Fotos von diesem Projekt.
Text: Biologie LK 2B7 2006; Fotos: Sören Rossmann; Blue Genes-Logo: Roche/Blue Genes
2006-04-03, sr